Section Abstract Introduction Methods Results Discussion Conflict of Interest Acknowledgment References
Clinical Research
Applicability of a clinical scoring criteria for disease severity of ß-thalassemia/ hemoglobin E in Indonesia
pISSN: 0853-1773 • eISSN: 2252-8083
https://doi.org/10.13181/mji.v27i1.1779 Med J Indones. 2018;27:26–32
Received: January 14, 2017
Accepted: January 8, 2018
Author affiliation:
1 Department of Child Health, Faculty of Medicine, Universitas Indonesia, Cipto Mangunkusumo Hospital, Jakarta, Indonesia
2 Thalassemia Research Center, Institute of Molecular Biosciences, Mahidol University, Bangkok, Thailand
3 Eijkman Institute for Molecular Biology, Faculty of Medicine, Universitas Indonesia, Cipto Mangunkusumo Hospital, Jakarta, Indonesia
Corresponding author:
Pustika A. Wahidiyat
E-mail: pa.wahidiyat@gmail.com
Background
β-thalassemia/HbE presents with a variety of clinical symptoms, from asymptomatic to severe, requiring routine transfusion. However, there is currently no agreed classification system to stratify patients based on clinical severity of β-thalassemia/HbE in the Indonesian population. Thailand has already established a classification system, and this study aimed to identify the applicability of the Thailand clinical scoring criteria to determine the severity of β-thalassemia/HbE in the Indonesian population.
Methods
This descriptive study was conducted by evaluating patients with β-thalassemia/HbE, who were classified into mild, moderate, or severe groups based on the Thailand clinical scoring criteria.
Results
A total of 293 subjects with β-thalassemia/HbE were included. Based on this clinical scoring criteria, it was found that only 21.5% of patients were classified as mild, and the remaining 35.5% and 44% were classified as moderate and severe respectively. Approximately 68.2% of the subjects in the severe group received transfusion at <4 years old, while only 10% of those in the mild group were transfused at the same age. In the mild group, only 10% of the subjects underwent routine transfusion, compared to 98.4% of the subjects in the severe group. In addition, only 27% of the subjects in the mild group showed stunted growth, while that in the moderate and severe groups were 54.5% and 86.8%, respectively.
Conclusion
Thailand clinical scoring criteria is able to determine the severity of Indonesia thalassemia patient which needs further management, i.e. transfusion and observation of stunted growth. This scoring system will help provide the provision of the most ideal management for the groups of patients based on their requirements.
Keywords
β-thalassemia/HbE, clinical criteria, Indonesia, scoring, Thailand
Thalassemia and other hemoglobinopathies are the most common causes of single gene disorders in the world, occurring due to the decrease of or the lack of synthesis of a-globin or b-globin chain (the primary component of adult hemoglobin; a2b2), which is inherited in an autosomal recessive pattern. The World Health Organization (WHO) stated that there are not less than 250 million people worldwide who are carriers of this disorder, corresponding to approximately 4.5% of the world population.1 Given its increasing prevalence, the appropriate diagnosis and severity classification for thalassemia also becomes increasingly important, in order to provide the most appropriate treatment for this condition.
The clinical classification for thalassemia is based on several factors, including age at first diagnosis, age at data collection, hemoglobin level at first diagnosis, and average hemoglobin level. If the patient receives routine transfusion, several other factors are also taken into account, including age at first transfusion, frequency of transfusion, presence of organomegaly, changes in bone structure, developmental disorders, and general condition of the patient.2 Based on the data above, thalassemia is further divided into mild, intermediate, or severe groups.3 A group of investigators in Thailand have devised a simpler clinical assessment system that involves 6 parameters, including average pre-transfusion hemoglobin, age at first diagnosis, age at first transfusion, spleen size, frequency of transfusion, and body height. In the world, there are several different scoring systems available, but these particular scoring criteria appear to be more applicable in the clinical setting.4
Even though patients have the same genetic mutation, the phenotypic clinical manifestations of patients with b-thalassemia/ HbE are extremely variable, which can range from asymptomatic patients that were coincidentally detected to those with severe symptoms requiring routine transfusion. However, regardless of this varying degree of severity for this disease, currently in Indonesia there is no standardized definition of when a patient is determined to be clinically mild or severe. Research has shown that Southeast Asian nations may share common ancestry, and the population share similar characteristic features.5 One clinical scoring criteria devised in Thailand, known as the Thailand clinical scoring criteria, has been established as a valid classification criteria to evaluate the disease severity of b-thalassemia/ HbE patients in the Thailand population, which has been found to be applicable in 950 subjects with b-thalassemia/HbE.4 This has prompted the investigators to devise a clinical scoring criteria for b-thalassemia/HbE patients in the Indonesian population.
Identifying the degree of severity for this disorder in the Indonesian population is important, so that the treatment can be tailored according to the degree of severity, which may then improve the quality of life of the patients. Often, we see that all patients diagnosed with b-thalassemia/HbE automatically receive transfusion, whereas in reality the spectrum of the disease is variable, and they do not always require transfusion. Providing the correct treatment for patients in accordance to the degree of severity will also minimize the risks of blood borne infections and delay the occurrence of iron overload. Following the rationale stated above, this study aimed to identify the applicability of a clinical criteria for patients with b-thalassemia/ HbE in the Indonesian population to determine the clinical severity of these patients.
METHODS
A cross-sectional study design was employed for this study. Patients with b-thalassemia/HbE (based on hemoglobin analysis) were identified and grouped based on their clinical manifestations into mild, intermediate, or severe using a clinical scoring system formulated by the Thalassemia Research Centre at Mahidol University, Thailand.4
The population of the study was all patients with b-thalassemia/HbE in Indonesia and the samples were all patients with b-thalassemia/HbE treated at the Thalassemia Centre, Department of Child Health and Department of Internal Medicine of RSUPN Dr. Cipto Mangunkusumo, Eijkman Institute for Molecular Biology, and affiliated hospitals in Indonesia from December 2006 to December 2008. The ethical clearance for this study was obtained from the Ethical Committee of the Faculty of Medicine, Universitas Indonesia, on 4th December 2006 (Letter No: 263/PT02.FK/ETIK/2006).
The inclusion criteria for the study were patients whose hemoglobin analysis indicated a diagnosis of b-thalassemia/HbE and were willing to participate in the study. Participants were excluded if they had a complication of the disorder or a severe infection.
Each participant or parent of the participant that fulfilled the inclusion criteria was provided with an informed consent form to be read and signed. They were also provided with explanation on the purpose and benefits of the study and details on the examinations. The data collected included demographic data, including gender and date of birth, clinical data, including body height, body weight, age at first diagnosis, age at first transfusion, frequency of transfusion, spleen size (in Schuffner and cm, evaluated using a ruled instrument to calculate the length of spleen portion palpable below the rib arc), and history of previous splenectomy, as well as laboratory data, including average pretransfusion hemoglobin in the last 3 years from the time of data collection and the hemoglobin level at first diagnosis.
Following data collection, the subjects were classified into mild, moderate, or severe based on the Thailand clinical criteria to determine the proportion of each severity group for b-thalassemia/HbE patients in Indonesia.
RESULTS
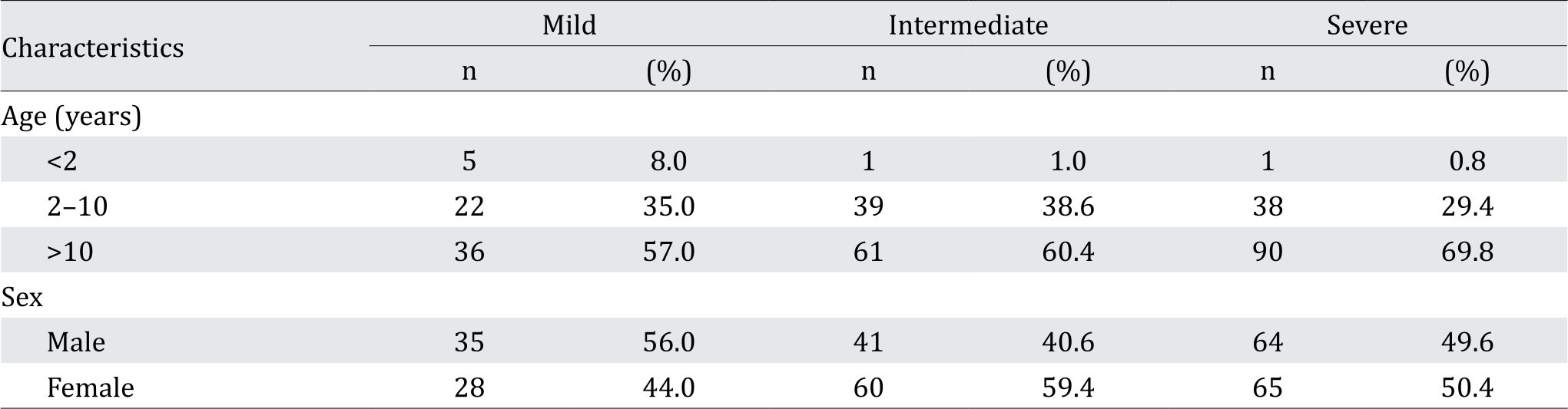
Following data collection, 293 subjects fulfilled the inclusion criteria and were classified into mild (63 subjects, or 21.5%), intermediate (101 subjects, or 35.5%), or severe (129 subjects, or 44%) group. The age of the subjects ranged from 1–38 years old. Table 1 shows the demographic characteristics of the study population.
Table 1. Demographic characteristics of the study population at screening of the study
Age at first diagnosis
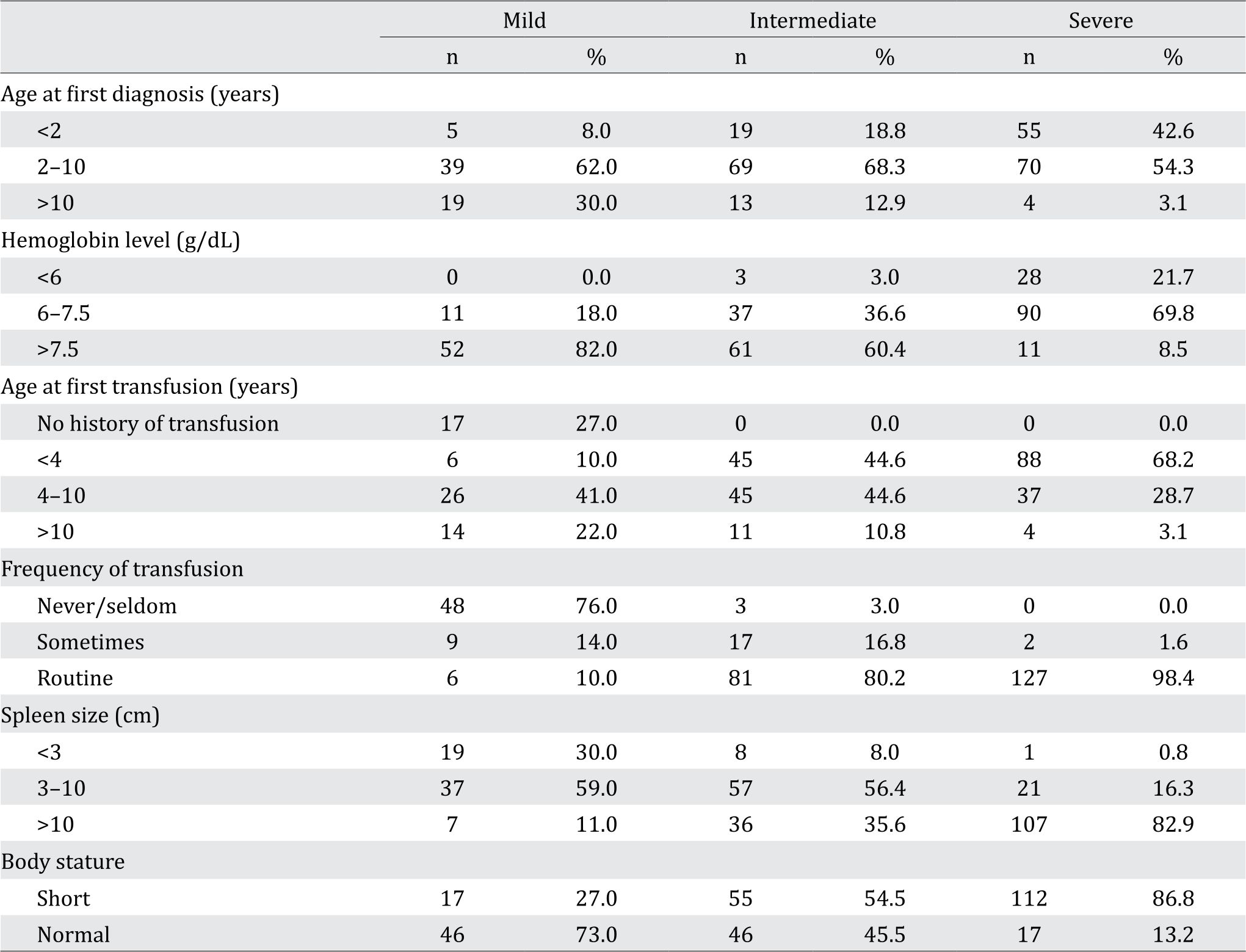
The results shown in Table 2 demonstrates that as the disease increases in severity at a younger age diagnosis.
Table 2. The distribution of the subjects based on the age at first diagnosis, age at first transfusion, average pre-transfusion hemoglobin level, frequency of transfusion, spleen size, and linear growth
Age at first transfusion
A majority of the subjects in the severe group generally had received their first transfusion before the age of 4 (68.2%), while only 4 (3.1%) subjects in this group received transfusion after the age of 10. In contrast, subjects in the mild group were more likely to receive transfusion at an older age, with 90% of the subjects receiving transfusion after the age of 4.
Average pre-transfusion hemoglobin
A large proportion of subjects in the mild and moderate groups had pre-transfusion hemoglobin of >7.5 g/dL, at 82.0% and 60.4%, respectively. On the other hand, 69.8% of the subjects in the severe group had a pretransfusion hemoglobin level of 6–7.5 g/ dL. The data for the average pre-transfusion hemoglobin in the three groups are shown in Table 2.
Frequency of transfusion
There were 48 (76%) subjects from the mild group who received blood transfusion at a maximum of 2 times/year, and there were 6 (10%) subjects who received transfusion between 5 to 12 times/year. A large majority of the subjects in the intermediate and the severe group required routine transfusion. Only 2 (1.6%) subjects from the severe group required transfusion at most 3 times/year.
Spleen size
Spleen size In the mild group, 7 (11%) subjects had a spleen size of >10 cm, of which 2 subjects had undergone splenectomy. Fifteen (23.6%) subjects in the mild group did not show any signs of spleen enlargement. In the intermediate group, 8 subjects have undergone splenectomy, and the spleen size for this group ranged from 1–27 cm. Most of the subjects in the severe group had a spleen size >10 cm and 15 of these (14.0%) had undergone splenectomy.
Linear growth
As the clinical manifestations increased in severity, the percentage of subjects with body height below the normal curve also increased, indicating stunted growth. It was found that the number of subjects with stunted growth was 17 (27%) for the mild group, 55 (54.5%) for the intermediate group, and 112 (86.8%) for the severe group. Table 2 summarizes the data for each of the six variables evaluated.
DISCUSSION
This study aimed to evaluate the applicability of the Thailand clinical scoring criteria to classify the severity of b-thalassemia/ HbE in the Indonesian population. Assessment of clinical manifestation is important as it corresponds to the treatment provided for the patient, who may require a specific and tailormade management that can vary among patients. Although the management of thalassemia in Indonesia is not yet uniform, significant improvements are observed compared to the past. Currently, in the Thalassemia Centre at the Department of Child Health of RSUPN Dr. Cipto Mangunkusumo, 33% of the patients are >18 years old.6 However, with the increasing age of the patients and acknowledgement of the fact that complications usually arise in the second decade, a comprehensive management involving several disciplines is required to ensure that the patients may achieve an optimal growth.
The results from this study classified 21.5% of the patients as mild, 35.5% as intermediate, and 44.0% as severe. This was slightly different to that from the Sripichai et al.4 study, which identified a relatively equal distribution between the three groups with 30% classified as mild, 37% as intermediate, and 33% as severe. One assumption is that the high incidence of co-inheritance between b-thalassemia/HbE and a-thalassemia causes the clinical signs and symptoms of the patient to be less severe, placing them in the mild and moderate groups.4 Some of the reasons that are thought to account for this may be related to the lack of early detection and awareness of the population regarding the signs and symptoms of thalassemia, as well as the lack of carrier testing.6
Many of the subjects (68.3%) in the intermediate group were initially diagnosed at ages 2 to 10. It was in accordance to other studies, which also found that the age at diagnosis for this group ranged from 2 to 8 years.7,8 From physical examination, the subjects in this study who were diagnosed after the age of 10 had already manifested severe complications, including changes in facial bone structure, delayed puberty, growth disorders, and splenomegaly. Similarly, a study from Thailand found that the subjects from the severe group who did not receive adequate transfusion may acquire complications in the first decade of life.9A delay in seeking treatment is often found in developing countries, which may be due to the lack of knowledge and awareness of the population on children’s health.10,11
Six subjects in the mild group received initial transfusion before the age of 4, which in retrospect could have been postponed as 3 did not require further transfusions. The provision of unnecessary transfusion, aside from increasing the financial costs incurred by the patient, may also affect the prognosis.12 In the intermediate group, a majority of the subjects (89.2%) received their initial transfusion at ages 4 to 10, in accordance with a study in Italy, in which subjects in the same group showed clinical manifestations after the age of 2.9 It is difficult to use "age at first transfusion" as a parameter in determining the severity of the patients, as there is no uniformity in the provision of blood transfusion in Indonesia, and it is highly dependent on the clinical assessment and decision of each medical professional.
The subjects had an average pretransfusion hemoglobin level of 7.3 g/dL (1.0 SD), with a range of 4.5-11 g/dL. This is in accordance with the population of patients with b- thalassemia in Thailand who had an average pre-transfusion hemoglobin of 7.7 g/dL and a range of 3.0-13.0 g/dL.13 In this study, the average hemoglobin level for the mild, intermediate, and severe groups were found to be 8.1 g/dL (0.7 SD), 7.5 g/dL (0.8 SD), and 6.6 g/dL (0.9 SD) respectively. Similarly, in the study conducted in Thailand, the average hemoglobin level for the mild, intermediate, and severe groups were found to be 8.2 g/dL (1.2 SD), 6.8 g/dL (1.2 SD), and 6.0 g/dL (1.0 SD) respectively.2
A majority of the subjects in the intermediate group required routine transfusion (80.2%), and only 3 subjects required a maximum of 2 times/year. This is quite different from a study by Camaschella et al.8, which found that 42% of intermediate thalassemia patients in Italy did not require transfusion. This discrepancy may be due to a difference in the type of mutation, clinical scoring criteria, and management of the patients. The majority of thalassemia patients in Indonesia often do not receive adequate transfusion. Moreover, the presence of hypersplenism will also accelerate the occurrence of hemolysis and anemia.9
The results indicated that the greater the spleen size, the more severe the clinical manifestations that appear. The proportion of subjects with a spleen size of <2 cm in the mild, intermediate, and severe groups were 30%, 8%, and 0.8% respectively, which is in accordance with the findings from the population in Thailand, which had a result of 39.1%, 10.5%, and 0.32% for the mild, intermediate, and severe groups respectively.4
The proportion of subjects with normal linear growth in the mild, intermediate, and severe groups were 73.0%, 45.5%, and 13.2% respectively. However, in the Thailand study from which the severity scoring was adapted, the proportion of subjects with normal linear growth in the mild, intermediate, and severe groups were 87.9%, 78.1%, and 43.5% respectively.4 There is a discrepancy between the Indonesian and Thailand population, especially in the proportion of subjects with normal growth in the intermediate and severe groups. This may be due to the difference in the growth chart used. Thailand has devised their own growth chart based on their population, and the anthropometric measurements for this study used the Centers for Disease Control and Prevention (CDC) 2000 growth chart, which is currently acceptable and widely used to evaluate the growth of Indonesian children.
The primary advantage of using a clinical scoring system to classify b-thalassemia/ HbE patients in Indonesia is that it can lead to improvements in determining treatment based on the severity of the disease. Specifically, the improvements that can be conducted include the provision of tailored iron chelation therapy and provision of blood transfusion based on the degree of clinical severity. Currently, a majority of b-thalassemia/HbE patients receive transfusion regardless of the disease severity. However, in hindsight, a majority of mild patients do not require routine transfusion. Therefore, implementing a scoring system to classify patients can be the first step to determine treatment strategies for each class of disease severity. However, a disadvantage of the scoring criteria is the discrepancy in terms of the instruments used for anthropometric measurements in the Thailand and Indonesian population.
The limitations of this study include the fact that regression analysis was not performed to evaluate which variables from the clinical scoring criteria were clinically significant in the Indonesian population due to time constraints. Furthermore, variables such as linear growth for the Indonesian population are still evaluated using the CDC 2000 growth chart, whereas many countries already have specific growth charts for their populations. The clinical implication of this study is that an improved treatment provision is expected to be conducted if a scoring criteria is used to determine disease severity, and this scoring criteria is recommended to be used in the Indonesian population. Further investigations are required to determine which other variables may influence the clinical manifestations of b-thalassemia/HbE in the Indonesian population in order to accurately classify the patients and provide the most ideal management.
In conclusion, using the Thailand clinical scoring system to evaluate the incidence of severity of b-thalassemia/HbE patients, 21.5% of the Indonesian patient population were classified as mild, whereas the remaining 35.5% and 44.0% were classified as moderate and severe respectively. Thailand clinical scoring criteria is able to determine the severity of Indonesia thalassemia patient which needs further management, i.e. transfusion and observation of stunted growth. This clinical scoring system will be beneficial in terms of improving the management strategies for b-thalassemia/HbE patients in Indonesia.
Conflicts of Interest
The authors affirm no conflicts of interest in this study.
Acknowledgment
The authors would like to thank Nitish Basant Adnani for his assistance and contributions in the preparation of this manuscript.
REFERENCES
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;88(6):480–7.
- Fucharoen S, Weatherall DJ. The hemoglobin E thalassemias. Cold Spring Harb Perspect Med. 2012;2(8):a011734.
- Danjou F, Anni F, Galanello R. Beta-thalassemia: from genotype to phenotype. Haematologica. 2011;96(11):1573–5.
- Sripichai O, Makarasara W, Munkongdee T, Kumkhaek C, Nuchprayoon I, Chuansumrit A, et al. A scoring system for the classification of beta-thalassemia/HbE diasease severity. Am J Hematol. 2008;83(6):482–4.
- Soares P, Trejaut JA, Loo JH, Hill C, Mormina M, Lee CL, et al. Climate change and postglacial human dispersals in Southeast Asia. Mol Biol Evol. 2008;25(6):1209–18.
- Wahidiyat PA. Thalassemia and its problems in Indonesia [Lecture]. Presented at: The 2nd international scientific meeting on hematology, oncology, thrombosis, and transplantation/transfusion in indonesia. Jakarta; 2016.
- Sahu PK, Pati SS, Mishra SK. Genotype-phenotype correlation of b-thalassemia spectrum of mutations in an Indian population. Hematol Rep. 2012;4(2):e9.
- Musallam KM, Taher AT, Rachmilewitz EA. b-Thalassemia intermedia: a clinical perspective. Cold Spring Harb Perspect Med. 2012;2(7):a013482.
- Fucharoen S, Winichagoon P. Clinical and hematological aspects of hemoglobin E beta-thalassemia. Curr Opin Hematol. 2000;7(2):106–12.
- Panigrahi I, Agarwal S. Genetic determinants of phenotype in beta-thalassemia. Hematology. 2008;13(4):247–52.
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12(2):61–76.
- Cappellini N, Cohen A, Eleftheriou A, Piga A, Porter J. Guidelines for the clinical management of thalassemia. Thalassemia International Federation; 2000.
- Sripichai O, Munkongdee T, Kumkhaek C, Svasti S, Winichagoon P, Fucharoen S. Coinheritance of the different copy numbers of alpha-globin gene modifies severity of beta-thalassemia/Hb E disease. Ann Hematol. 2008;87(5):375–9.
Copyright @ 2018 Authors. This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original author and source are properly cited.
mji.ui.ac.id